헬리코박터 제균 치료가 위암 연관 유전자의 후성유전학적 변화에 미치는 영향
Effect of Helicobacter pylori Eradication on Epigenetic Changes in Gastric Cancer-related Genes
Article information

Trans Abstract
It is known that gastric carcinogenesis results from the progressive changes from chronic gastritis to gastric atrophy, intestinal metaplasia, dysplasia, and invasive carcinoma. Several genetic and epigenetic alterations are involved in this process, and Helicobacter pylori (H. pylori) infection is believed to induce the initiation and progression of these steps. From an epigenetic point of view, H. pylori induces hypermethylation of genes involved in the development of gastric cancer and regulates the expression of various microRNAs (miRNAs). These H. pylori-related epigenetic changes are accumulated not only at the site of neoplasm but also in the adjacent non-cancerous gastric mucosa. Thereby, a state vulnerable to gastric cancer known as an epigenetic field defect is formed. H. pylori eradication can have an effective chemopreventive effect in gastric carcinogenesis. However, the molecular biological changes that occur in the stomach environment during H. pylori eradication have not yet been established. Several studies have reported that H. pylori eradication can restore infection-related changes, especially epigenetic alterations in gastric cancer-related genes, but some studies have shown otherwise. Simply put, it appears that the recovery of methylated gastric cancer-related genes and miRNAs during H. pylori eradication may vary among genes and may also differ depending on the histological subtype of the gastric mucosa. In this review, we will discuss the potential mechanism of gastric cancer prevention by H. pylori eradication, mainly from an epigenetic perspective.
서 론
지난 수십 년간 위암의 발생률이 감소하고 있지만, 2020년 기준으로 위암은 여전히 전 세계 다섯 번째로 많이 발생하는 암이며, 네 번째로 흔한 암 사망 원인으로 꼽힌다[1]. 특히 동아시아 지역에서 발생률이 높아 전 세계 위암 환자의 3/4이 아시아인이며, 한국에서는 위암이 전체 암 발생률 1위를 차지한다[2].
최근의 역학 연구에 따르면 우리나라 성인의 약 51.0%가 Helicobacter pylori (H. pylori)에 감염되어 있다고 보고되었다. H. pylori는 위점막에 수십 년에 걸쳐 만성 염증을 일으키며, 시간이 경과함에 따라 위축성 위염, 장상피화생, 이형성의 단계를 거쳐 위암으로 진행할 수 있어 세계보건기구 규정 1급 발암물질로 인정된다[3-5]. H. pylori가 위암을 일으키는 기전에 대해서는 다양한 가설이 있는데, 염증 관련 면역세포의 비정상적인 활성화 및 사이토카인의 증가 유도, 종양억제유전자(tumor suppressor gene)의 비활성화 또는 종양유전자(oncogene)의 활성화, 위암 줄기세포(gastric cancer stem cell)의 생성 유도 등이 제시되고 있다[6]. 이 중, H. pylori에 의한 위암 관련 유전자들의 발현 변화는 DNA의 돌연변이(mutation) 등에 의한 유전자 자체의 변화보다 후성유전학적 변화(epigenetic change), 즉 DNA 염기서열에 아무런 변화가 생기지 않으면서 유전자 발현 패턴이 영향을 받는 경우가 더 많은 것으로 보고된다[7]. 후성유전학적 변화에는 본 논고에서 주로 다룰 DNA 메틸화(DNA methylation) 연구가 가장 잘 알려져 있으며, 그 외에도 염색질(chromatin)의 기본 단위인 뉴클레오솜(nucleosome)을 구성하는 히스톤 단백질에 아세틸화, 메틸화, 인산화, 유비퀴틴화 등의 변형이 일어나면서 뉴클레오솜의 구조에 영향을 미치는 히스톤 변형(histone modification), 단백질을 암호화하지 않는 small RNA의 일종으로, 표적 mRNA와 결합하여 단백질로의 번역(translation)을 억제하거나 분해시킴으로써 유전자 발현을 제어하는 microRNA (miRNA) 및 응축된 염색질을 열리거나 닫히게 하여 유전자 발현을 조절하는 염색질 재형성(chromatin remodeling) 등 여러 형태의 조절 기전이 있다[6].
한편, 지금까지 H. pylori 제균 치료가 위암의 발생을 줄일 수 있는가에 대한 많은 연구들이 있어 왔다. 그 결과 H. pylori 감염의 초기 단계에서는 제균에 따른 위암 발생 억제 효과가 비교적 뚜렷하게 입증되었으나, 위축성 위염이나 장상피화생과 같은 전암성 변화가 이미 진행된 후에는 제균 치료를 통한 위암의 예방 효과가 뚜렷하지 않다는 소위 “point of no return” 가설이 지배적이었다[8-10]. 그러나 최근에는 이러한 전암성 변화를 가진 환자들에서도 제균 시 위암 발생률 감소를 보이는 연구 결과가 누적되면서 제균 치료의 고려 대상이 점차 확대되고 있다[11-14]. 다만, H. pylori의 성공적인 제균 후에도 여전히 위암은 발생할 수 있어, 제균 후의 위 내 환경의 회복에는 가역적 및 비가역적인 영역이 혼재되어 있을 것으로 추측된다[15]. H. pylori 제균 치료가 구체적으로 위 내의 환경을 어떻게 변화시키며, 어떠한 분자생물학적 기전을 통해 위암의 발생을 억제하는지에 대해 많은 기초 연구가 있어왔지만 현재까지 명확한 결론을 도출하기는 어려운 상태이다.
본 논고에서는 H. pylori 제균 치료 이후 위 내 환경의 분자 생물학적 회복, 그중에서도 특히 대표적인 후성유전학적 기전인 DNA 메틸화 및 miRNA의 발현 양상의 변화에 대해 살펴봄으로써, 위암의 화학예방(chemoprevention)에 있어 제균 치료가 가지는 의의를 되새겨보고자 한다.
본 론
1. H. pylori 감염에 따른 위암 관련 유전자의 DNA 메틸화 변화
후성유전학적 변화는 거의 모든 종류의 암의 발생과 관련 있는 중요 기전으로, 그중 가장 대표적인 것이 DNA 메틸화이다[16]. 인간 유전자 촉진자(promoter)의 약 60~70%에 CpG dinucleotide (Cytosine-Guanine이 phosphate 하나로 연결된 부위)가 수백에서 수천 개 모여 있는 CpG island가 발견되는데, DNA 메틸화는 이 CpG island에 위치한 cytosine의 5번 탄소에 메틸기가 결합하여 5-methylcytosine으로 변화하는 생화확적 변화이다. 암이 발생하는 과정에서는 크게 2가지의 비정상적인 메틸화 양상이 관찰되는데, 유전자 촉진자 내의 CpG island에서 발생하는 DNA 과메틸화(regional hypermethylation)와 유전체(genome) 전반적으로 메틸화가 오히려 감소하는 저메틸화(global DNA hypomethylation)가 그것이다[16,17]. 종양억제 유전자 촉진자의 DNA 과메틸화는 촉진자 영역에 전사인사(transcription factor)의 결합을 방해함으로써 해당 유전자의 발현을 억제하고, 유전체 전반의 저메틸화는 DNA 구조의 불안정성을 가중시킴으로써 암 발생을 유도한다(Fig. 1) [18,19].
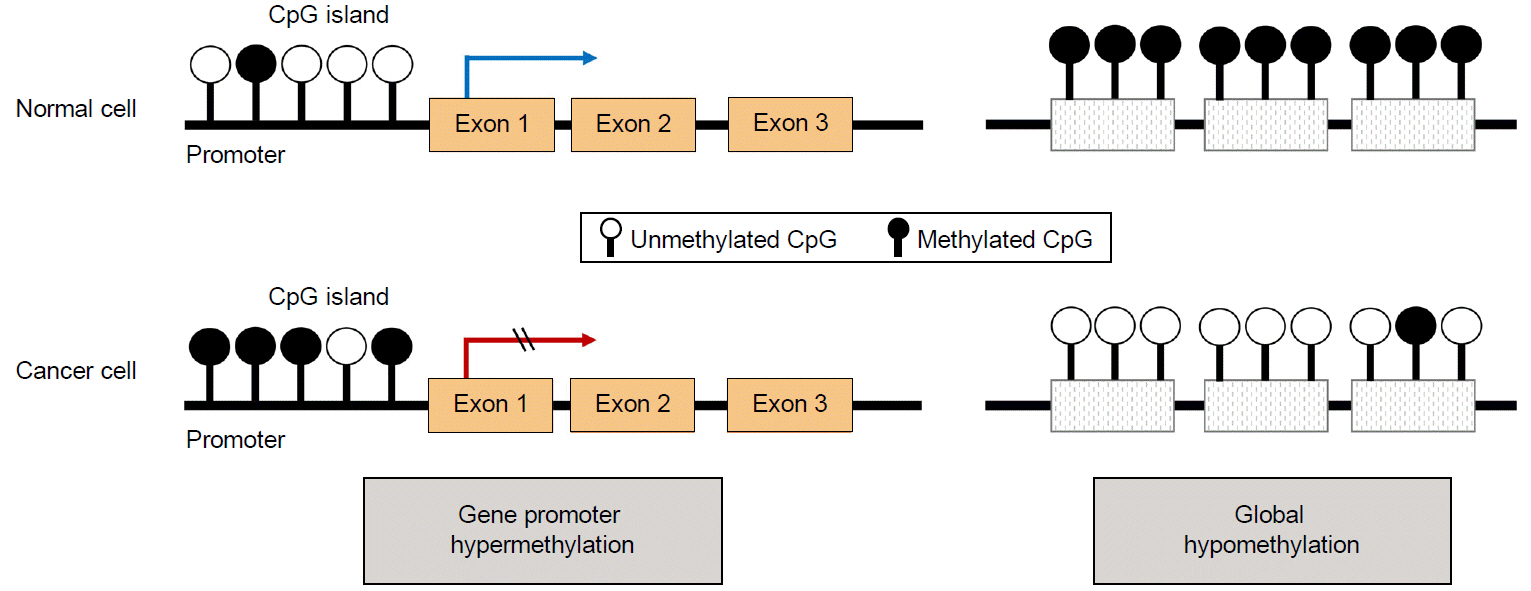
DNA methylation in cancer. Cancer cells show both hypo- and hypermethylation events. Global loss of DNA methylation at repeated sequences contributes to genomic instability and activation of oncogenes. Moreover, DNA hypermethylation at specific sites, especially in the promoter region of tumor suppressor genes, results in silencing of the corresponding gene.
H. pylori 감염자의 위 점막에서 E-cadherin 유전자(CDH1)가 과메틸화된다는 결과가 처음 발표된 이후로 현재까지 H. pylori 감염이 p16, MLH1, LOX, RUNX3 등의 다양한 종양억제유전자의 촉진자 영역에서 DNA 메틸화를 유도한다고 보고되었으며, 이를 통해 해당 유전자들의 발현을 억제함으로써 위암 발생을 유도하는 것으로 설명한다(Table 1) [20-54]. 한편으로 H. pylori 감염은 정상적으로는 메틸화되어 있던 인체 유전자의 반복 염기 서열인 ALU와 LINE-1 등을 탈메틸화하여 유전체 전반의 저메틸화를 초래하는 경로를 통해서도 위암 발생 과정에 기여한다[55]. H. pylori 감염으로 인한 이러한 유전자의 비정상 메틸화는 전암성 병변인 위축성 위염, 장상피화생 및 이형성증 단계에서도 발생되며, 이에 따라 H. pylori 양성 위암 환자의 암 주변 정상 위점막에서도 비정상 메틸화가 증가되어 있다[32,56,57]. 즉, 장기간의 H. pylori 감염에 위 전반이 지속적이고 광범위하게 노출되고 이에 따라 위암 관련 유전자들의 DNA 메틸화 변화가 누적됨으로써, 노출된 점막의 어느 곳에서나 여러 단계의 전암성 병변 및 위암이 다발적으로 발생할 수 있는 “epigenetic field for cancerization”이 형성된다는 가설이 중요한 개념으로 받아들여지고 있다(Fig. 2).
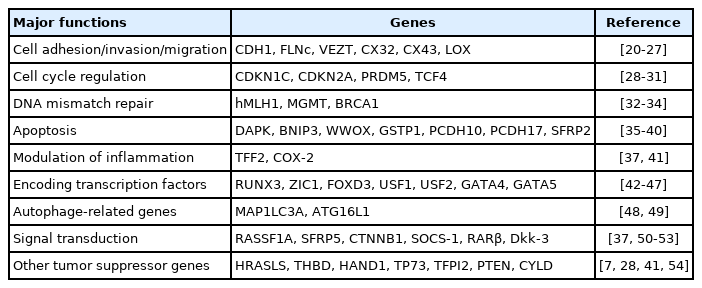
Genes Regulated by Helicobacter pylori-induced DNA Methylation in Gastric Carcinogenesis
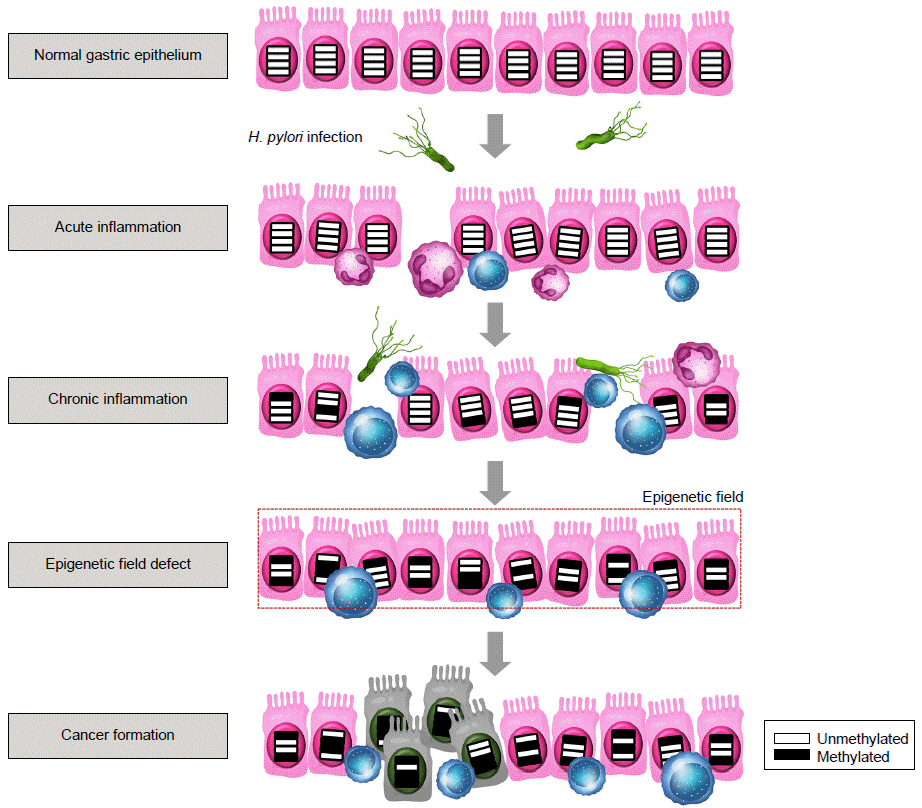
Epigenetic field defect by Helicobacter pylori (H. pylori) infection. H. pylori infection causes acute infiammation, followed by chronic inflammation if the infection persists. Consequently, chronic infiammation triggered by H. pylori induces aberrant DNA methylation in the gastric mucosa. Accumulation of these epigenetic changes form an "epigenetic field for cancerization", which plays a key role in the development of gastric cancers.
한편 H. pylori 감염으로 인한 DNA 메틸화의 정도는 세균이 가지는 독성인자(virulence factor)에 따라 차이를 보인다고 보고되어 있다. H. pylori 독성인자와 관련하여 여러 유전자형이 존재하는데, 그중 cag pathogenicity island는 cagA 유전자를 포함하는 약 40 kb의 DNA insertion element이다[58]. cagA 유전자에서는 CagA 단백을 생성하는데, 서구의 H. pylori 균주는 약 60~70%에서 CagA 양성인 반면, 한국을 포함한 동아시아에서는 거의 100% CagA 양성 균주로 보고된다[59]. CagA 양성 H. pylori 균주는 CagA 음성 균주에 비해 심한 염증 반응을 초래하며, 임상적으로는 심한 위축성 위염, 이형성 및 위 선암의 발생 위험도를 높인다는 연구 결과들이 있어왔다[60-63]. 균주의 CagA 존재 유무에 따른 DNA 메틸화를 분석해 본 연구에서 CagA 양성 균주에 감염되었을 시 음성 균주에 감염된 경우보다 유전자들의 메틸화 정도가 높음을 알 수 있었는데, 특히 DNA 복구(DNA repair)를 담당하는 단백질인 O6-methylguanine DNA methyltransferase를 암호화하는 유전자 및 위암 발생을 억제하는 기능을 하는 miRNA인 let-7의 촉진자 영역에 과메틸화가 관찰되어 H. pylori 감염이 위암을 일으키는 기전에 대한 하나의 단서로 작용할 수 있겠다[64].
H. pylori 감염이 어떤 기전을 통해 위암 관련 유전자의 메틸화 수준을 변화시키는지는 아직 완전히 이해되지는 못하였으나, 만성 염증(chronic inflammation)이 그 과정에 중요한 역할을 할 것으로 생각되고 있다. H. pylori가 위 점막을 감염시키면 IL-1β, IL-6, IL-8, TNF-α와 같은 염증성 사이토카인의 분비가 증가하고 다양한 염증 세포 침윤이 야기된다[65,66]. 또한 반응 산소 종(reactive oxygen species)과 반응 질소 종(reactive nitrogen species) 등의 산화 스트레스가 유발됨으로써 DNA 메틸화 효소인 DNA methyltransferase의 활성도가 증가하게 된다[67]. Mongolian gerbil 동물 모델을 이용한 in vivo 실험에서 H. pylori에 감염된 개체에 cyclosporin A를 투약하여 위 점막 내의 H. pylori 수는 유지하면서 염증 반응을 억제하였을 때 DNA의 비정상 메틸화가 감소함을 볼 수 있었는데, 이는 일단 H. pylori 감염에 의해 염증이 시작되고 나면 이후로는 H. pylori 균 자체보다 H. pylori에 의해 유발된 염증이 DNA의 이상 메틸화에 중요하다는 점을 간접적으로 제시한다[68].
2. H. pylori 감염에 따른 miRNA의 발현 변화
H. pylori 감염에 따른 후성유전학적 변화로 최근 각광받는 또 하나의 분야는 miRNA이다[69]. miRNA는 DNA로부터 전사(transcription)가 되지만 단백질로는 번역되지 않는 20-24 염기 서열의 짧은 RNA 조각으로, 표적 mRNA에 상보적으로 결합하여 단백질의 합성을 억제하거나 mRNA의 분해를 유도하는 방식을 통해 target 유전자의 발현을 전사 후 수준에서 조절(posttranscriptional regulation)한다(Fig. 3). 현재까지 약 1,000여 개의 miRNA가 보고되었으며, 부분적 상호보완성으로도 mRNA에 결합할 수 있어 하나의 miRNA가 약 200개의 protein-coding gene을 조절할 것으로 예측된다[70,71]. miRNA는 종양억제유 전자 또는 종양유전자의 전사 후 조절에도 관여함으로써 세포의 증식(proliferation), 세포 자멸사(apoptosis) 등에 영향을 미치며 이를 통해 위암을 비롯한 다양한 암의 발생 및 진행과 연관되어 있음이 알려졌다[72]. 현재까지 H. pylori 연관 위암 환자에서 발현이 변화하는 100여 개의 miRNA가 발견되었는데, 연구 결과들이 누적되면서 이들 miRNA가 위암에서 진단, 치료 및 예후 예측의 잠재적 바이오마커로서 역할을 할 것으로 기대되고 있다[73].

Mechanisms of microRNA (miRNA)-mediated post-transcriptional regulation. miRNA, which is a short, non-coding RNA, is generated through the following process: (1) RNA polymerases type II (Pol-II) generates a long primary precursor miRNA (pri-miRNA), (2) nuclear RNase III enzyme Drosha cleaves pri-miRNA to release precursor miRNA (pre-miRNA), (3) pre-miRNA is exported out of the nucleus to the cytoplasm by exportin-5 receptor, (4) the cytoplasmic RNase III enzyme Dicer cleaves the pre-miRNA to form miRNA duplex containing the mature miRNA, and (5) the duplex unwinds and the mature miRNA incorporates into RNA-induced silencing complex (RISC). miRNA acts as a post-transcriptional regulator by targeted mRNA degradation or translation inhibition.
H. pylori 연관 위암 발생 과정에서 miRNA deregulation이 어떠한 역할을 하는지 아직 완전히 알려지지 않았지만, 대표적으로 miR-17, miR-20a, miR-21, miR-146a, miR-155, miR-223 등은 H. pylori 감염 및 위암 환자에서 발현이 증가되어 있어 종양유전자로 기능할 것으로 예측되며, let-7 family, miR-31, miR-101, miR-125a, miR-141, miR-203, miR-204, miR-210, miR-218, miR-335, miR-375, miR-377, miR-449 등은 그 발현이 감소되어 있어 종양억제유전자로 기능할 것으로 예측된다[74]. 세부 연구에서 miR-21 과발현은 PDCD4, TIMP3, PTEN, TPM1, RHOB 등의 종양억제유전자 발현 억제를 통해 종양 세포의 증식, 세포 자멸사, 침윤(invasion), 혈관신생(angiogenesis)에 관여하고, miR-375 발현 감소는 PDK1, JAK2 등의 종양유전자를 활성화시켜 세포의 증식, 이동(migration), 침윤을 유도하며, miR-204 발현 감소는 snai1 및 SOX4 유전자를 targeting함으로써 상피간엽이행(epithelialme-senchymal transition)을 가속화하는 것으로 보고되었다[75-80].
그렇다면 H. pylori 감염은 어떠한 기전을 통해 이러한 miRNA의 발현을 변화시킬까? 현재까지 알려진 몇 가지 기전은 다음과 같다. 우선 miRNA 또한 protein-coding RNA와 마찬가지로 전사인자에 의해 조절될 수 있다. 즉, 전사인자가 miRNA의 촉진자 영역에 결합하면 pre-miRNA의 전사를 촉진하고 이는 mature miRNA의 발현을 증가시킨다. 예를 들어, H. pylori의 병원성 단백질인 CagA는 MYC의 발현을 증가시키고, 이는 miR-17-92 cluster의 regulatory region에 결합함으로써 miR-17-92 cluster를 활성화시킬 수 있다[81,82]. 다음으로 miRNA의 발현 조절에도 앞서 살펴본 후성유전학적 조절이 관여하는 것으로 보인다[69,83,84]. 인간 miRNA의 약 1/3이 상류 영역(upstream region)에 CpG island를 가지고 있으며, DNA 메틸화에 의해 발현이 조절된다고 추정된다[85]. 이전 연구를 살펴보면 위암 환자에서 miR-10b, miR-124, miR-137, miR-210, miR-129-2 등이 촉진자 메틸화 기전을 통해 발현이 감소되며, 이는 target 유전자인 MAPRE1, SMOX, CDC42, STMN1, SOX4를 과발현 시킨다고 보고되어 있다(Table 2) [69,77,86-120]. 또한 위암 세포주를 대상으로 한 연구에서 DNA demethylation agent인 5-AZA-2'-deoxycytidine (5-AZA-CdR)이나 histone deacetylase inhibitor인 4-phenylbutyric acid (PBA)를 처리하였을 때 이상 조절된 miRNA의 발현이 회복됨이 보고되었다. 위암 세포주에 5-AZA-CdR 처리 시 NOTCH4와 KRAS 유전자를 표적으로 하는 miRNA-181c의 발현이 증가하였고, miR-34b/c 또한 5-AZA-CdR 및 PBA 처리 시 발현이 증가되었다[96,106]. 본 저자들의 연구에서도 위암 세포주에 5-AZA-CdR 처리 시 miR-133a 및 miR-200a/b의 발현이 증가함을 보고한 바 있다[102,112]. 5-AZA-CdR 및 PBA 처리로 miRNA 발현이 회복될 수 있다는 사실은 후성유전학적 기전이 miRNA의 발현에 관여함을 시사한다.
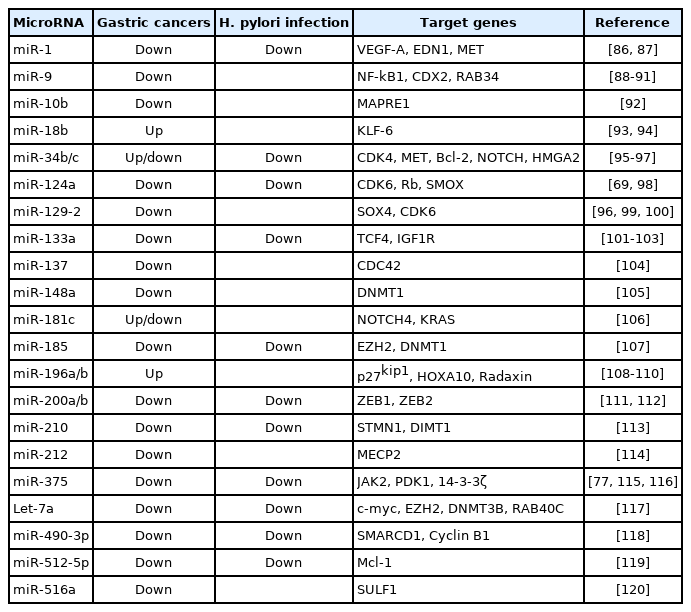
MicroRNAs Regulated by DNA Methylation in Gastric Cancers and Helicobacter pylori Infection
3. H. pylori 제균 치료가 DNA 메틸화 및 miRNA의 회복에 미치는 영향
H. pylori 감염은 급·만성 위염에서 시작하여 위축성 위염, 장상피화생, 이형성을 거쳐 위암에 이르기까지 장기간에 걸쳐 위 점막에 직간접적인 영향을 주기 때문에 단연코 제균 치료는 가장 확실한 위암 발생 억제책으로 기대된다. 과거부터 진행되어온 임상 연구에서는 감염의 지속 기간에 따라 제균 치료를 통한 위암 억제 효과의 차이를 보여 왔는데, 위축성 위염이나 장상피화생과 같은 변화가 진행된 후에는 제균 효과가 뚜렷하지 않은, 즉 제균 치료의 “point of no return”으로 인식되어 왔다. 하지만 비교적 최근의 연구들에서는 이러한 point of no return을 넘어선 환자들에서도 제균 후 위암의 발생이 감소하는 결과들이 다수 보고되어, 전암성 변화를 가진 군에서의 제균 효과 및 이를 토대로 한 제균 치료의 대상 선정에 있어서 아직 논란이 끝나지 않은 상태이다[11-14]. 기초 연구에 있어서도 H. pylori 감염으로 인해 위 내에 형성된 epigenetic field가 제균 시 구체적으로 어떠한 분자생물학적 변화를 겪게 되는지에 대해 많은 연구자들이 실험적 증거를 제시하고 있으나 그 변화의 범위 및 가역성 여부에 대해서는 아직까지 명확한 결론이 도출되지 못한 상태이다.
제균 치료를 통한 위암 관련 유전자의 후성유전학적 변화의 가역성을 본 연구로는 다음의 연구가 대표적이다. CDH1은 세포 부착과 종양 억제 기능을 하는 E-cadherin을 코딩하는 유전자로, 가족성 및 산발성 위암 모두에서 E-cadherin의 silencing이 관찰된다[20]. 앞서 언급한 바와 같이 H. pylori 감염 시 CDH1 유전자의 촉진자 영역 메틸화가 증가하는데, 제균 치료 후 해당 유전자의 메틸화 정도가 유의하게 감소되었다[21]. 또 다른 연구에서도 H. pylori 제균 치료 시 p16, APC, COX2의 촉진자 메틸화 정도가 낮아졌으며, DNA 손상을 복구하는 유전자인 O6-methylguanine DNA methyltransferase의 촉진자 메틸화 정도도 제균 후 유의하게 감소하였다[32,33]. 다만 같은 연구에서 MLH1 유전자의 메틸화 정도는 제균 전후에서 유의한 차이를 보이지 않았다[32]. miRNA를 대상으로 제균 치료 전후의 촉진자 메틸화 변화를 비교한 연구들에서도 후성유전학적 변화가 유의하게 호전된 결과들이 보고되었다. 본 저자들의 연구실에서는 종양 억제 기능을 가진 것으로 알려진 miRNA인 miR-133a의 촉진자 메틸화 정도가 대조군, H. pylori 양성 위염, H. pylori 양성 위암 환자군으로 갈수록 증가하며, H. pylori 양성군에서 제균 치료 시 촉진자 메틸화 정도가 유의하게 감소함을 발표하였다[102]. 또한, miR-200a와 miR-200b를 대상으로 한 연구에서도 제균 치료 후 miRNA 유전자 메틸화 수준이 유의하게 호전됨을 확인하였다[112]. miR-200a와 miR200b는 miR-200 family의 구성원으로, 상피간엽이행을 유발하는 전사인자인 zinc finger E-box binding homeobox (ZEB1/ZEB2)를 억제함으로써 여러 암종의 발생 과정에 핵심적인 단계를 조절하는 것으로 알려져 있다[121]. 본 연구에서 miR-200a/b의 촉진자 메틸화 정도는 대조군, H. pylori 양성 위염, H. pylori 양성 위암 환자 순으로 증가하였고, 제균 치료 시 H. pylori 양성군에서 촉진자 메틸화 정도가 유의하게 감소하는 소견이 발견되었다. 상기의 연구 결과들은 H. pylori 감염으로 인해 형성된 epigenetic field for cancerization이 제균 치료를 통해 회복시킬 수 있으며, 이것이 제균 치료 후 위암 발생이 감소하는 하나의 기전이 될 수 있음을 시사하였다.
한편, 제균 이후에도 메틸화가 유의하게 호전되지 않음을 보인 연구 결과들도 존재한다. 제균 치료 후 장기간의 추적 관찰 기간(평균 26.0개월)을 보유한 연구를 살펴보면 LOX 유전자의 메틸화 정도는 유의하게 감소하였으나 APC 유전자는 유의한 차이를 보이지 않았고, MOS는 동반된 조직학적 변화 정도에 따라 메틸화 회복 여부에 차이를 보여 제균 후 메틸화 회복 여부는 gene-specific manner를 따랐다[122]. 해당 연구에서 MOS 유전자의 경우 장상피화생이나 이형성 등의 전암성 병변이 동반되지 않은 군에서는 제균 이후 메틸화 정도가 감소하는 반면, 전암성 병변 또는 위선암이 동반된 환자의 위 점막에서는 제균 치료에도 불구하고 메틸화 정도가 감소하지 않았다. 또한 본 저자들의 연구실에서 수행한 연구에서도 제균 치료 후 위암 관련 유전자들 중 p16, CDH1의 메틸화 정도는 감소하였으나 RUNX-3의 메틸화 정도에는 유의한 호전이 관찰되지 않았으며, Wnt 길항체 유전자들(SFRP1, -2, -5, DKK1, -2, -3, WIF1)을 대상으로 한 연구에서는 제균 치료 후 7개의 Wnt 길항체 유전자 중 오직 DKK3에서만 DNA 메틸화가 감소된 소견을 발견하였다[123,124]. 나아가 제균 치료 후 후성유전학적 회복 효과를 위 점막의 조직학적 아형에 따라 나눠 살펴본 연구들에서는 흥미로운 결과가 관찰되었다. 협대역 영상(narrow band imaging)을 통해 위 점막의 미세 구조(microarchitecture)를 restored type과 atrophic type으로 구별할 수 있는데, 이 중 atrophic type은 조직학적으로 장상피화생과 밀접한 관련이 있다고 알려져 있다[125]. 제균 치료 후 miR-124a의 발현은 restored type에 비해 atrophic type에서 유의하게 낮았는데, 이는 miR-124a-3의 CpG 과메틸화와 연관성이 있었다. 다른 연구에서는 laser capture microdissection 등의 기법으로 위 점막을 세포 수준에서 장상피화생 및 비장상피화생으로 나누어 제균 치료의 효과를 분석하였는데, 위암군에서 발현이 증가된 oncogenic miRNAs (miR-17/92, miR-106b-93-25, miR-21, miR-194, miR-196)가 장상피화생에서는 제균 치료 후에도 이상 발현이 회복되지 않음이 관찰되었다[126]. miR-124a-3의 메틸화 회복에 대해 살펴본 다른 연구에서도 장상피화생에서는 제균 후 해당 유전자의 메틸화가 호전되지 않음이 보고된 바 있다[127]. 즉, 제균 치료 후에도 일부 유전자들의 후성유전학적 변화는 호전되지 않을 수 있으며 이는 특히 장기간의 H. pylori 감염으로 인해 장상피화생이 진행된 부분에서 제균 치료 후에도 변화가 비가역적으로 지속되는 것이 원인일 수 있을 것으로 추측된다. 향후 연구를 통해 H. pylori 제균 치료의 분자 생물학적 기전에 대한 추가적인 이해와, 이를 토대로 제균 치료 후 위암 발생의 위험도를 예측할 수 있는 표지자를 발굴함으로써 임상 현장에서의 활용이 기대되는 바이다.
결 론
H. pylori 제균 치료는 강력한 위암 억제 효과를 나타내지만, 제균 치료 후에도 분명히 위암 발생의 위험성은 남아있다. 본 논고에서 살펴본 바와 같이 기초 연구들을 통해 제균 치료 후 위 내 환경의 분자생물학적 회복 기전에 대해 많은 부분을 설명할 수 있게 되었으나, 임상 현장에서 성공적인 제균 치료 후에도 위암의 발생 위험성이 높은 개인을 선별하여 추적 관찰하기 위한 유용한 바이오마커의 개발이 여전히 요구된다.
Notes
No potential conflict of interest relevant to this article was reported.